
Etiquetas
#pandemia #COVID-19 #triaje #toma de decisiones #comorbilidades #cuidados paliativos #Dolor crónico #Neurocirugía #Síndrome de Fracaso de Cirugía Espinal Lumbar #anestesia espinal unilateral #dosis mínima #lateralización #cirugía traumatológica ambulatoria #nervio pudendo #neuralgia #radiofrecuencia pulsada; ultrasonido. #dolor #dolor neuropatico #topiramato #dolor agudo postquirúrgico #dolor incisiona #anestesia #niños #dolor agudo #dolor quirurgico #modelos y teorias #enfermeria #noticias #Investigación en Servicios de Salud #Rehabilitación #ansiedad #emociones #miedo #tristeza #Parche #Buprenorfina #Fentanilo #problemas sociales #síndrome pediátrico inflamatorio multisistémico #fibromialgia #narrativas #experiencia #Afrontamiento #estados emocionales #dolor de espalda #Cuidado paliativo #opioidesRevista El Dolor 50 | Diciembre 2007 - Año 17 | Colaboraciones Especiales
Neuroplasticidad y Dolor
Cerveró, Fernando (*)
Unidad de Investigación de Anestesia y Centro McGill para la Investigación del Dolor
McGill University, Montréal, Québec, Canadá
Resumen
Todas Ias formas de doIor incIuyen eI desarroIIo de un estado de hiperaIgesia que iIustra Ia naturaIeza dinámica y pIástica de Ia sensación de doIor. La hiperaIgesia es Ia característica más importante deI proceso doIoroso y es Ia expresión de Ia hipersensibiIidad de Ias vías deI doIor inducida por Ia sensibiIización de Ios receptores periféricos que registran eventos doIorosos y de Ias neuronas que transmiten y procesan esta información sensoriaI aI SNC. Los nociceptores periféricos se sensibiIizan adquiriendo una mayor y a veces nueva capacidad de respuesta a Ios estímuIos periféricos. Por otra parte, un proceso de pIasticidad sináptica, deI cuaI se ha identificado una variedad de componentes moIecuIares, interviene en Ia ampIificación centraI de Ias señaIes de Ias aferencias nociceptivas, Io cuaI evoca Ia hipersensibiIidad de Ias neuronas centraIes. EI resuItado finaI es un proceso sensoriaI que, a pesar de haber sido puesto en marcha iniciaImente por una Iesión, puede no mantener una reIación estrecha con Ia Iesión originaI y convertirse en un estado de doIor crónico sin tener una causa definida.
Palabras Clave: DoIor, HiperaIgesia, AIodinia, NeuropIasticidad, SensibiIización, Nociceptor, Neuropatía
Abstract
AII forms of pain incIude the deveIopment of a hyperaIgesic state that iIIustrates the dynamic and pIastic nature of pain sensation. HyperaIgesia is the most prominent feature of the pain process and is the expression of hypersensitivity of the pain pathway induced by the sensitization of the peripheraI receptors that signaI painfuI events and of the neurons that transmit and process this sensory information to the CNS. PeripheraI nociceptors can be sensitized, acquiring enhanced, and sometimes noveI, responsiveness to peripheraI stimuIi. On the other hand a process of synaptic pIasticity, of which severaI moIecuIar components have aIready been identified, mediates the centraI ampIification of the afferent signaIs that Ieads to the hypersensitivity of centraI neurons. The finaI resuIt is a sensory process that, aIthough initiaIIy triggered by injury, may not keep a cIose reIationship with the originating injury and deveIop into a chronic pain state in the absence of a defined cause.
Keywords: Pain, HyperaIgesia, AIIodynia, NeuropIasticity, Sensitization,
Nociceptor, Neuropathy
Introducción
Siempre han existido dos visiones alternativas respecto al significado biológico del dolor. Una de ellas propone que el dolor es un sentido similar a la vista o al oído, un componente del repertorio sensorial que posee la mayoría de los animales que nos advierte sobre el peligro inminente, le entrega información precisa a nuestro cerebro sobre lesiones y nos ayuda a sanar. Sin embargo, siempre ha existido una interpretación alternativa del dolor que niega que éste sea como la vista o el oído, y propone que los papeles fundamentales tanto del dolor como de su opuesto, el placer, son la formación de emociones y comportamientos del individuo. El dolor se ve como el instrumento que gatilla estados emocionales, un motor del comportamiento y una herramienta de aprendizaje muy efectiva. Aristóteles, quien dio vida a esta interpretación, lo dejó muy claro: sólo hay cinco sentidos, la vista, el oído, el olfato, el gusto y el tacto. El dolor y el placer no son sentidos, sino "pasiones del alma".
La primera interpretación dio origen a la teoría de la Especificidad
del Dolor, que propone que los terminales nerviosos finos de aferentes amielínicos son receptores de dolor en la periferia y que existe una vía transmisora específica de dolor que lleva sus señales al cerebro. Esta teoría es responsable del muy conocido modelo de mecanismos de dolor que, a menudo, se encuentra en los libros y de acuerdo al cual un receptor de dolor en la periferia se ve activado por un estímulo nocivo y envía impulsos a la médula espinal y desde ahí al tálamo y la corteza por medio de una vía espino-talámico cruzada. En otras palabras, una vía de dolor relativamente simple y directa (Figura 1).
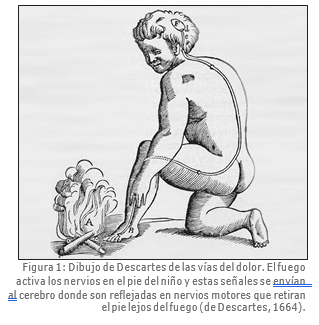
Se ha podido establecer con cierto grado de certeza que existen sensores especializados en la piel, músculos y vísceras en la mayoría de los animales que se activan, exclusivamente, por estímulos que causan lesiones y cuya excitación evoca la sensación de dolor (Belmonte y Cerveró, 1996). También sabemos que no se puede evocar un dolor, en circunstancias normales, cambiando los patrones de activación de los receptores sensoriales táctiles (Ochoa y Torebjörk, 1983, 1989). También existe una gran cantidad de datos que demuestran la existencia de neuronas en la médula espinal y el cerebro activadas, mayoritaria o exclusivamente, por estímulos nociceptivos (Hunt y Mantyh, 2001). Sin embargo, también hay una gran cantidad de evidencia experimental a favor de la plasticidad en el canal sensorial nociceptivo y de la existencia de procesos dinámicos que pueden alterar profundamente las propiedades funcionales de los nociceptores periféricos y de las neuronas centrales nociceptivas (Treede y otros, 1992; Julius y Basbaum, 2001; Hunt y Mantyh, 2001). También se ha demostrado que con posterioridad a una lesión o inflamación periférica, la activación de aferentes táctiles de la piel lesionada, puede evocar sensaciones de dolor (ver Cerveró y Laird, 1996).
¿Un dolor o varios dolores?
Hoy sabemos que los distintos tipos de dolor están producidos por mecanismos heterogéneos que participan en diversas formas en la generación de los estados dolorosos (Cerveró y Laird, 1991; Klein y otros, 2005). En general, podemos identificar tres formas distintas de dolor considerando la relación entre los estímulos nocivos y la sensación de dolor: nociceptivo, inflamatorio y neuropático (también llamados dolores fase 1, 2 y 3 por Cerveró y Laird, 1991). El dolor nociceptivo tiene relación con el procesamiento de los estímulos nocivos breves; el dolor inflamatorio es el resultado de una estimulación nociva prolongada que produce daño tisular, y el dolor neuropático se produce como consecuencia del daño neurológico, incluyendo las neuropatías periféricas y estados de dolor central (Figura 2).
El dolor nociceptivo es una sensación protectora necesaria para
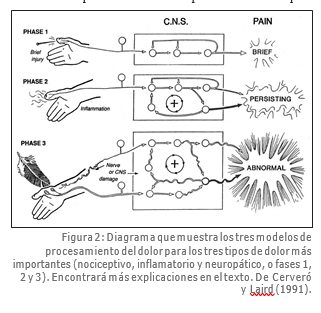
la supervivencia y el bienestar de los seres humanos. Los mecanismos que median el procesamiento de los estímulos nocivos breves pueden entenderse como una vía sencilla que transmite impulsos en los nociceptores periféricos hacia el tálamo y la corteza y genera una breve percepción de dolor. Por otra parte, una lesión y daño en los tejidos evocan una reacción inflamatoria como parte del proceso de cicatrización y generan un estado de dolor distinto al dolor nociceptivo producido por cambios en las propiedades de respuesta de los diversos componentes del sistema nociceptivo. Estos cambios incluyen la sensibilización del nociceptor y el reclutamiento de poblaciones de receptores que previamente no presentaban respuesta. A su vez, las neuronas del SNC muestran una amplificación en su capacidad de excitación expresada como aumentos en el tamaño del campo receptor de las neuronas y una mayor descarga espontánea y evocada. Todos estos cambios indican que el SNC se encuentra ahora en un nuevo estado, con mayor capacidad de excitación como resultado del estímulo nocivo generado por los tejidos lesionados o inflamados. En estas condiciones, se pierde la correlación inmediata entre las descargas en los nociceptores periféricos y la percepción de dolor.
Los síndromes de dolor neuropático son la consecuencia del
daño a los nervios periféricos o al mismo SNC y producen sensaciones de dolor fuera del rango de las sensaciones producidas por el sistema nociceptivo normal. Estas experiencias dolorosas incluyen dolor espontáneo, umbrales de dolor muy reducidos y dolor que se evoca simplemente con el tacto. Los estados de dolor neuropático se caracterizan por una casi completa falta de correlación entre los estímulos periféricos nocivos y la sensación de dolor y se producen a partir de lesiones neurológicas que causan una actividad anormal generada en los neuromas de los nervios lesionados o en las células de los ganglios de la raíz dorsal, así como conducción efáptica de impulsos entre fibras nerviosas adyacentes y respuestas anormales de los nociceptores periféricos y las neuronas del SNC. Los dolores nociceptivos e inflamatorios son síntomas de una lesión periférica, en tanto que el dolor neuropático es un síntoma de una patología neurológica. Si bien la teoría de la Especificidad puede explicar bastante bien las formas más simples de dolor tales como un pinchazo o dolor agudo a causa de una quemadura leve, la experiencia de dolores complejos, tales como el dolor provocado por estímulos de baja intensidad, requiere en gran medida de plasticidad periférica y central. Es muy probable que sistemas nociceptivos, tanto específicos como no específicos, participen en la generación y mantención de los distintos estados de dolor.
Plasticidad e hiperalgesia
´
El dolor es un proceso dinámico que no se puede explicar con una sola teoría o un mecanismo único. Una de las expresiones más llamativas de la naturaleza dinámica de la sensación de dolor es la falta de adaptación. Un estímulo continuo y uniforme, ya sea visual o auditivo, nos lleva a la adaptación sensorial; después de algunos segundos o minutos, simplemente dejamos de sentir este estímulo. Sin embargo, la sensación de dolor no sólo no se adapta a un estímulo nocivo y continuo, sino que se hace cada vez peor y al cabo de unos minutos de estimulación persistente donde el estímulo de dolor es relativamente suave, la sensación se hace insoportable. Este cambio en la sensibilidad al dolor genera un estado de amplificación del dolor o hiperalgesia, que es normalmente puesto en marcha y mantenido por un estímulo nocivo persistente, pero que puede, bajo circunstancias patológicas, aparecer sin una causa obvia, de manera que la relación normal entre una lesión y el dolor se pierde. La hiperalgesia y la alodinia son los síntomas más importantes de muchos estados de dolor crónico y las propiedades de la sensación de dolor que la hacen especialmente desagradable e insoportable. En términos psicológicos, un estado de hiperalgesia está representado por una desviación hacia la izquierda, inducida por estímulos dolorosos persistentes, en la curva que relaciona la intensidad del estímulo con la sensación de dolor (Cerveró y Laird, 1996). Este giro hace que la parte más baja de la curva caiga en el rango de intensidad inocuo del estímulo (alodinia o dolor producido por un estímulo inocuo), mientras que la parte superior muestra una sensación amplificada de dolor a estímulos nocivos (hiperalgesia o una mayor sensibilidad al dolor ante estímulos nocivos). La alodinia y la hiperalgesia ofrecen al organismo mecanismos de protección, previniendo que la persona estimule un área lesionada y, al mismo tiempo, ayudando en el proceso de curación (Figura 3).
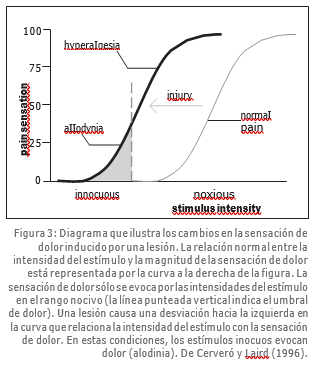
Existen dos tipos de hiperalgesia: primaria y secundaria. La hiperalgesia primaria se define como una mayor sensibilidad al dolor que ocurre en el lugar donde se produce la lesión. La hiperalgesia secundaria se define como una mayor sensibilidad al dolor que ocurre en áreas adyacentes o incluso remotas al lugar donde se produce la lesión. Por ejemplo, después de sufrir una lesión en una mano, se puede desarrollar un área de hiperalgesia que cubra todo el brazo, o la inflamación del intestino o la vejiga pueden producir un área de hiperalgesia en la región abdominal o pélvica.
Actualmente podemos identificar tres procesos clave en el desarrollo de los mecanismos de hiperalgesia: i) el proceso de sensibilización del nociceptor responsable de las señales iniciales de una lesión y los cambios periféricos en el sistema nociceptivo inducidos por estímulos nocivos, ii) el proceso de amplificación central de las señales nociceptivas (conocida como sensibilización central), generadas por el fortalecimiento sináptico de las conexiones entre las neuronas del SNC y responsable de la mayor capacidad de excitación que acompaña los estados de dolor persistente, y iii) el proceso por el cual la actividad en los receptores sensoriales de umbral bajo de áreas periféricas no lesionadas, puede acceder al sistema nociceptivo y evocar sensaciones de dolor y estados de hiperalgesia (dolor evocado por el tacto, alodinia táctil, etc.) (Figura 4).
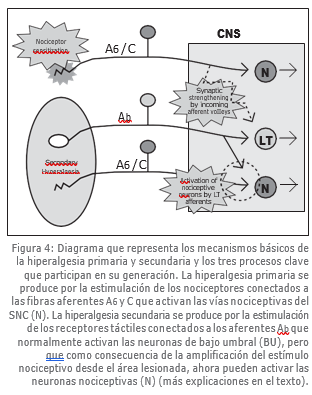
Mecanismos de hiperalgesia primaria: sensibilización de los nociceptores
Se sabe desde hace mucho tiempo que las propiedades funcionales de los nociceptores periféricos cambian como consecuencia de una lesión o inflamación en el territorio que éstos inervan (Treede y otros, 1992). Como resultado de esta propiedad conocida como sensibilización, los nociceptores, después de un periodo de activación intensa, adquieren una mayor capacidad de excitación ante un estímulo posterior. Disminuye su umbral y aumentan sus respuestas, mostrando una desviación hacia la izquierda en su función estímulo - respuesta, que encaja perfectamente con la desviación hacia la izquierda de la hiperalgesia sensorial. En algunos casos, la inflamación o una lesión pueden activar poblaciones de nociceptores que, con anterioridad, no presentaban
respuesta alguna (los así llamados nociceptores "silenciosos"). Esto se interpreta como una forma extrema de sensibilización del nociceptor.
Se cree que la sensibilización de los nociceptores es el mecanismo responsable de la hiperalgesia primaria. Aún no hemos entendido en su totalidad el proceso de sensibilización, pero en él participan factores titulares liberados por los propios terminales nociceptores o por células cercanas (como por ejemplo, los mastocitos). Como resultado de la hiperexcitabilidad del nociceptor en el lugar donde se produce la lesión, el umbral de dolor disminuye y aumentan las respuestas al dolor evocadas desde el área lesionada, esto es, se produce hiperalgesia primaria.
Mecanismos de hiperalgesia secundaria: sensibilización central
El hecho que la hiperalgesia secundaria aparezca en partes del cuerpo lejanas del lugar donde se produjo la lesión original, nos indica que el mecanismo mediador en esta forma de hiperalgesia es el sistema nervioso central y no una alteración periférica. Sabemos que los impulsos generados en los nociceptores en la zona lesionada o inflamada evocan cambios centrales que, a su vez, producen una hiperalgesia secundaria (LaMotte y otros, 1991; Torebjörk y otros, 1992). Estos cambios están disparados por los impulsos que llegan desde los nociceptores que inervan la zona lesionada y se mantienen debido a la mayor actividad espontánea de estos nociceptores a causa de su sensibilización. Este proceso se ha llamado sensibilización central por analogía a la sensibilización periférica observada en el lugar donde ocurre la lesión, e implica un aumento en la capacidad de excitación de las neuronas de segundo orden en la médula espinal y en las zonas supraespinales, y un cambio radical en las consecuencias sensoriales de la activación de las fibras aferentes de bajo umbral que va desde el tacto al dolor.
Datos experimentales demuestran que la actividad intensa o persistente en los nociceptores periféricos conduce a una mayor capacidad de respuesta de las neuronas del asta dorsal de la médula espinal (ver referencias en Cerveró y Laird, 1996). Este aumento en la capacidad de excitación de las neuronas de la médula espinal, coincide con el típico giro hacia la izquierda de la curva de dolor observada en humanos y animales de laboratorio en áreas de hiperalgesia secundaria, y es lógico pensar que un proceso lleva al otro. Por lo tanto, la idea de un proceso de sensibilización central ha sido aceptada como un mecanismo neural posible para la generación de estados de hiperalgesia. Se ha propuesto a muchos candidatos moleculares como mediadores de la sensibilización central, incluyendo a los neurotransmisores como el glutamato y los neuropéptidos, por ejemplo, la Sustancia P. Sin embargo, existen también muchos aspectos de este proceso que aún no son conocidos, incluyendo la relación entre el aumento en la capacidad de excitación observada en las neuronas de la médula espinal luego de recibir un estímulo nocivo y los cambios sensoriales específicos observados en los estados de hiperalgesia secundaria. Incluso, no está claro el significado exacto de la sensibilización central, ya que esta expresión se ha utilizado para calificar incremento en la respuesta al dolor en humanos y animales de laboratorio. Por otra parte, hoy sabemos que los procesos celulares que subyacen estos aumentos de excitabilidad son en todo similares a otras alteraciones plásticas de transmisión sináptica que se observan en regiones del cerebro no relacionadas al procesamiento del dolor. Es posible que la sensibilización central sea una expresión de la plasticidad sináptica en el sistema nervioso central y no un fenómeno específicamente relacionado al dolor.
Mecanismos de la alodinia: dolor evocado por el tacto
Las características de la hiperalgesia secundaria incluyen no sólo un aumento en la magnitud de las sensaciones de dolor evocadas desde esta zona, sino también un cambio en la modalidad de la sensación evocada por los mecanorreceptores de bajo umbral que cambia de tacto a dolor (Cerveró y otros, 1993; LaMotte y otros, 1991). Este proceso se conoce como dolor evocado por el tacto o alodinia táctil. En áreas de hiperalgesia secundaria, este dolor está generado por impulsos en los mecanorreceptores de bajo umbral conectados a fibras aferentes mielínicas de gran diámetro. Esto muestra que es posible producir dolor activando mecanorreceptores de bajo umbral, si bien en circunstancias especiales donde previamente haya ocurrido una lesión. El dolor evocado por el tacto puede aparecer muy poco tiempo después de haber activado los nociceptores desde el área primaria y puede también desaparecer muy rápidamente si se reduce o bloquea la actividad de estos nociceptores. El proceso es dinámico, reversible y no depende de la formación de nuevas conexiones anatómicas entre el sistema táctil y el nociceptivo.
Si bien se podría explicar la mayor sensibilidad al dolor en áreas
de hiperalgesia secundaria por una mayor capacidad de excitación del sistema nociceptivo, el cambio radical desde el tacto hasta el dolor en las acciones centrales de los mecanorreceptores de bajo umbral, requiere explicaciones alternativas. Se han propuesto varias hipótesis que abordan esta cuestión. Todas se basan en una reversión de las acciones de los neurotransmisores inhibitorios GABA y glicina, de manera que se convierten en moléculas excitatorias y facilitan la estimulación de bajo umbral a las neuronas nociceptivas (Price y otros, 2005).
Uno de los mecanismos propuestos se basa en el proceso conocido como Despolarización Aferente Primaria, un proceso generado en los terminales de las aferentes nociceptivas por la actividad de las fibras sensoriales de bajo umbral. Se sabe que el mediador en este caso es el GABA que genera esta despolarización debido a que dichos terminales poseen una concentración intracelular de iones de cloruro mayor que la normal. Cuando estas despolarizaciones son lo suficientemente intensas como para evocar potenciales de acción, se abre una vía directa para que las fibras sensoriales de bajo umbral activen las aferentes nociceptivas y, por ende, la vía nociceptiva.
Otro mecanismo propuesto se basa en la desinhibición de la vía que conecta las aferencias de bajo umbral con las neuronas nociceptivas. En condiciones normales, esta vía está inhibida por interneuronas locales GABA-érgicas, pero la liberación de esta inhibición puede descubrir una conexión aferente de bajo umbral del sistema nociceptivo.
Finalmente, se ha propuesto otro mecanismo basado en la total reversión de las acciones de GABA o glicina en las neuronas de la médula espinal, desde la inhibición a la excitación (Coull y otros, 2003). Esto podría ser posible por el aumento en la concentración intracelular de los iones de cloruro en estas neuronas como consecuencia de la inhibición del co-transportador responsable de reducir el cloruro interno. En estas circunstancias, GABA y glicina se transforman en transmisores excitatorios, induciendo la despolarización de las neuronas, y no su hiperpolarización. Como consecuencia, las acciones inhibitorias normales de las aferentes de bajo umbral sobre las neuronas nociceptivas se convierten en elementos de excitación.
Conflicto de Intereses
Correspondencia
Fernando Cerveró
Unidad de Investigación de Anestesia McGiII University
McIntyre MedicaI BIdg. Room 1207 3655 Promenade Sir WiIIiam OsIer MontreaI, Quebec H3G 1Y6 CANADA
TeIéfono: +1 (514) 398-5764
FAX +1 (514) 398-8241
fernando.cervero@mcgiII.ca
Referencias Bibliográficas
1. BeImonte C, Cervero F. 1996. NeurobioIogy of nociceptors (NeurobioIogía de Ios nociceptores) pp. 1-531. Oxford: Oxford University Press
2. Cervero F, Laird JMA. 1991. One pain or many pains?: a new Iook at pain mechanisms (¿Un doIor o muchos doIores?: una nueva visión de Ios mecanismos de doIor) News PhysioI. Sci. 6: 268-73
3. Cervero F, Laird JMA. 1996. Mechanisms of touch-evoked pain (aIIodynia): A new modeI Pain 68: 13-23 (Mecanismos de doIor evocados por eI tacto (aIodinia): Un nuevo modeIo DoIor 68: 13-23)
4. Cervero, F., GiIbert, R., Hammond, R.G.E. y Tanner, J. 1993. DeveIopment of secondary hyperaIgesia foIIowing non-painfuI thermaI stimuIation of the skin: a psychophysicaI study in man. (DesarroIIo de hiperaIgesia secundaria posterior a una estimuIación no doIorosa de Ia pieI: estudio psicoIógico en eI hombre.) Pain 54: 181-189.
5. CouII JA, Boudreau D, Bachand K, Prescott SA, NauIt F, Sik A, De Koninck P, De Koninck Y. 2003 Trans-synaptic shift in anion gradient in spinaI Iamina I neurons as a mechanism of neuropathic pain. (Desviación trans sináptica deI gradiente en aniones en Ias neuronas de Ia Iámina I de Ia méduIa espinaI como mecanismo deI doIor neuropático.) Nature, 424: 938-942.
6. Descartes R. L'homme. Paris: Chez Jacques Le Gras, 1664.
7. Hunt SP, Mantyh PW. 2001. The moIecuIar dynamics of pain controI (La dinámica moIecuIar deI controI deI doIor) Nature Reviews Neuroscience 2: 83-91
8. JuIius D, Basbaum AI. 2001. MoIecuIar mechanisms of nociception (Mecanismos moIecuIares de Ia nocicepción) Nature 413:203-10
9. KIein T, MagerI W, RoIke R, Treede RD. 2005. Human surrogate modeIs of neuropathic pain (ModeIos humanos de reempIazo deI doIor neuropático) Pain 115: 227-33
10. LaMotte, R.H., Shain, C.N., Simone, D.A. y Tsai, E.-F.P. 1991 Neurogenic hyperaIgesia: PsychophysicaI studies of underIying mechanisms, (HiperaIgesia neurogénica: Estudios psicoIógicos de Ios mecanismos subyacentes) J. NeurophysioI. 66: 190-211.
11. Ochoa J, Torebjörk E. 1983. Sensations evoked by intraneuraI microstimuIation of singIe mechanoreceptor units innervating the human hand (Sensaciones evocadas por Ia microestimuIación intraneuraI de unidades mecano receptoras que inervan Ia mano humana) J. PhysioI 342: 633-54
12. Ochoa J, Torebjörk E. 1989. Sensations evoked by intraneuraI microstimuIation of C nociceptor fibres in human skin nerves (Sensaciones evocadas por Ia micro estimuIación intraneuraI de Ias fibras de nociceptores C en Ios nervios de Ia pieI humana) J. PhysioI 415: 583-99
13. Price T.J., Cervero F. y de Koninck Y. 2005 RoIe of Cation-ChIoride-Cotransporters (CCC) in pain and hyperaIgesia (RoI de Ios Co transportadores de Cationes de CIoruro (CCC) en eI doIor y Ia hiperaIgesia) Current Topics in MedicinaI Chemistry 5: 547-555.
14. Torebjörk, H.E., Lundberg, L.E.R. y LaMotte, R.H. 1992 CentraI changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperaIgesia in humans, (Cambios centraIes en eI procesamiento de información de mecanoreceptores en Ia hiperaIgesia inducida por capsaicina en humanos) J. PhysioI. (Lond.), 448: 765-780.
15. Treede R-D, Meyer RA, Raja SN, CampbeII JN. 1992. PeripheraI and centraI mechanisms of cutaneous hyperaIgesia (Mecanismos periféricos y centraIes de Ia hiperaIgesia cutánea) Prog. NeurobioI. 38: 397-421
versión impresa
ISSN 0717-1919Patrocinios



